The ONIOM scheme (Our owN n-layered Integrated molecular Orbital + molecular mechanics Method) was (is being) developped in Morokuma group (Emory University). The method is implemented in Gaussian 98 for instance. The ONIOM scheme is mainly derived from IMOMO, and further informations can be found in the reference to this methods (S. Humbel, S. Sieber and K. Morokuma, J. Chem. Phys. 105 (1996) 1959-1967.).
There are a number of other similar hybrid methods such as Nancy's QM/MM method based on localized orbitals rather than a link atom.
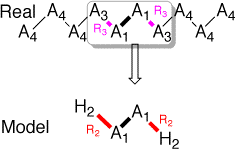 Briefly stated, the method is based on a selection (model system) of a reactive part in a large system, that is cutted out. In the model system, the outer part is replaced by link atoms (usually Hydrogens) located at a distance noted R2. In this special issue of Theochem on QM/MM type of methods, we use an harmonic approximation to analyze the ONIOM dependency toward this link atom distance, also called R2. In the gaussian implementation (as of g98), R2 is made proportional to the distance of the atom the hydrogen replaces (R3) through a factor (g).
Briefly stated, the method is based on a selection (model system) of a reactive part in a large system, that is cutted out. In the model system, the outer part is replaced by link atoms (usually Hydrogens) located at a distance noted R2. In this special issue of Theochem on QM/MM type of methods, we use an harmonic approximation to analyze the ONIOM dependency toward this link atom distance, also called R2. In the gaussian implementation (as of g98), R2 is made proportional to the distance of the atom the hydrogen replaces (R3) through a factor (g).
R2 = g R3 (1)
(1)
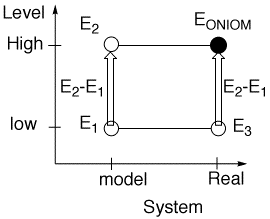
The ONIOM energy is a simple equation:
EONIOM = E3 + E2 - E1 (2)
where
E3 = Energy of the large system at a low level
E2 = Energy of the model system at a High level
E1 = Energy of the model system at a low level
The harmonic approximation 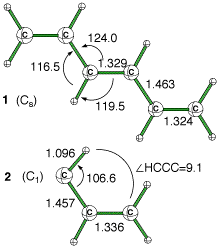 concerns the energy variation of model systems computation ( Ei = E2 or E1) as a function of R2. These energy are suposed to vary with quadratic variation (with an equilibrium geometry for the link atom (R20,i = x0,i) and a force constants (ki).
concerns the energy variation of model systems computation ( Ei = E2 or E1) as a function of R2. These energy are suposed to vary with quadratic variation (with an equilibrium geometry for the link atom (R20,i = x0,i) and a force constants (ki).
On the basis of a few example such as conjugated double bonds breaking, it is shown in this paper that the ONIOM absolute energy does not depend on the choice of this distance. To this statement it must be add "when an appropriate low level is used" , and "except for clearly 'unreasonable' distances". A strategy to analyze the low level is proposed on the basis of this harmonic approximation. 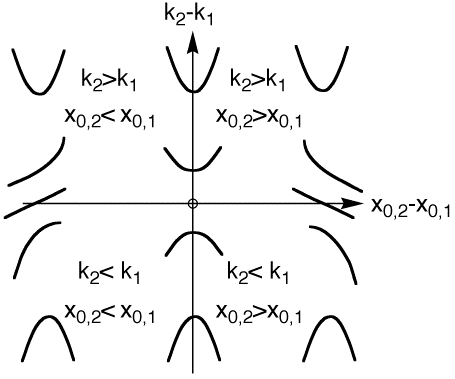 Geometry optimizations are found even less sensitive to the choice of this link atom distance.
Geometry optimizations are found even less sensitive to the choice of this link atom distance.
The small dependency of the ONIOM scheme toward the choice of the R2 distance for both the energy and the geometry supports the choice implemented in Gaussian (Equ. 1). However, special care would be welcome when the embedded methods bear a very different equilibrium geometry for the link atom (x0,i) and/or very different force constants (ki).
